EVAPORATION
Evaporation is a type of
vaporization of a
liquid, that occurs only on the
surface of a liquid. The other type of vaporization is
boiling, that instead occurs on the entire mass of the liquid. Evaporation is also part of the water cycle.
Evaporation is a type of
phase transition; it is the process by which
molecules in a
liquid state (e.g.
water) spontaneously become
gaseous (e.g.
water vapor). Generally, evaporation can be seen by the gradual disappearance of a liquid from a substance when exposed to a significant volume of gas.
Vaporization and evaporation however, are not entirely the same processes.
On average, the molecules in a glass of water do not have enough heat energy to escape from the liquid. With sufficient heat, the liquid would turn into vapor quickly (see
boiling point). When the molecules collide, they transfer energy to each other in varying degrees, based on how they collide. Sometimes the transfer is so one-sided for a molecule near the surface that it ends up with enough energy to escape.
Liquids that do not evaporate visibly at a given temperature in a given gas (e.g. cooking oil at room temperature) have molecules that do not tend to transfer energy to each other in a pattern sufficient to frequently give a molecule the heat energy necessary to turn into vapor. However, these liquids
are evaporating, it's just that the process is much slower and thus significantly less visible.
Evaporation is an essential part of the
water cycle.
Solar energy drives evaporation of water from
oceans,
lakes,
moisture in the soil, and other sources of water. In
hydrology, evaporation and
transpiration (which involves evaporation within
plant stomata) are collectively termed
evapotranspiration. Evaporation is caused when water is exposed to air and the liquid molecules turn into water vapor which rises up and forms clouds.
Theory
For
molecules of a liquid to evaporate, they must be located near the surface, be moving in the proper direction, and have sufficient
kinetic energy to overcome liquid-phase intermolecular forces.
[1] Only a small proportion of the molecules meet these criteria, so the rate of evaporation is limited. Since the kinetic energy of a molecule is proportional to its temperature, evaporation proceeds more quickly at higher temperatures. As the faster-moving molecules escape, the remaining molecules have lower average kinetic energy, and the temperature of the liquid thus decreases. This phenomenon is also called evaporative cooling. This is why evaporating
sweat cools the human body. Evaporation also tends to proceed more quickly with higher flow rates between the gaseous and liquid phase and in liquids with higher
vapor pressure. For example, laundry on a clothes line will dry (by evaporation) more rapidly on a windy day than on a still day. Three key parts to evaporation are heat, humidity and air movement.
On a molecular level, there is no strict boundary between the liquid state and the vapor state. Instead, there is a
Knudsen layer, where the phase is undetermined. Because this layer is only a few molecules thick, at a macroscopic scale a clear phase transition interface can be seen.
Evaporative equilibrium

Vapor pressure of water vs. temperature. 760
Torr = 1
atm.
If evaporation takes place in a closed vessel, the escaping molecules accumulate as a
vapor above the liquid. Many of the
molecules return to the liquid, with returning molecules becoming more frequent as the
density and
pressure of the vapor increases. When the process of escape and return reaches an
equilibrium,
[1] the vapor is said to be "saturated," and no further change in either
vapor pressure and density or liquid temperature will occur. For a system consisting of vapor and liquid of a pure substance, this equilibrium state is directly related to the vapor pressure of the substance, as given by the
Clausius-Clapeyron relation:
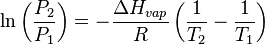
where
P1,
P2 are the vapor pressures at temperatures
T1,
T2 respectively, Δ
Hvap is the
enthalpy of vaporization, and
R is the
universal gas constant. The rate of evaporation in an open system is related to the vapor pressure found in a closed system. If a liquid is heated, when the vapor pressure reaches the ambient pressure the liquid will
boil.
The ability for a molecule of a liquid to evaporate is largely based on the amount of
kinetic energy an individual particle may possess. Even at lower temperatures, individual molecules of a liquid can evaporate if they have more than the minimum amount of kinetic energy required for vaporization.
Factors influencing the rate of evaporation
- Concentration of the substance evaporating in the air
- If the air already has a high concentration of the substance evaporating, then the given substance will evaporate more slowly.
- Concentration of other substances in the air
- If the air is already saturated with other substances, it can have a lower capacity for the substance evaporating.
- Concentration of other substances in the liquid (impurities)
- If the liquid contains other substances, it will have a lower capacity for evaporation.
- Flow rate of air
- This is in part related to the concentration points above. If fresh air is moving over the substance all the time, then the concentration of the substance in the air is less likely to go up with time, thus encouraging faster evaporation. This is the result of the boundary layer at the evaporation surface decreasing with flow velocity, decreasing the diffusion distance in the stagnant layer.
- Inter-molecular forces
- The stronger the forces keeping the molecules together in the liquid state, the more energy one must get to escape.
- Pressure
- In an area of less pressure, evaporation happens faster because there is less exertion on the surface keeping the molecules from launching themselves.
- Surface area
- A substance which has a larger surface area will evaporate faster as there are more surface molecules which are able to escape.
- Temperature of the substance
- If the substance is hotter, then its molecules have a higher average kinetic energy, and evaporation will be faster.
- Density
- The higher the density, the slower a liquid evaporates.
In the US, the National Weather Service measures the actual rate of evaporation from a standardized "pan" open water surface outdoors, at various locations nationwide. Others do likewise around the world. The US data is collected and compiled into an annual evaporation map.
[2] The measurements range from under 30 to over the120 inches (3,000 mm) per year.
Applications
When
clothes are hung on a laundry line, even though the ambient temperature is below the boiling point of water, water evaporates. This is accelerated by factors such as low
humidity,
heat (from the sun), and
wind. In a
clothes dryer hot air is blown through the clothes, allowing water to evaporate very rapidly.
Combustion vaporization
Fuel
droplets vaporize as they receive heat by mixing with the hot gases in the combustion chamber. Heat (energy) can also be received by radiation from any hot refractory wall of the combustion chamber.
CRYSTALLIZATION
Crystallization is the (natural or artificial) process of formation of solid
crystals precipitating from a
solution,
melt or more rarely
deposited directly from a
gas. Crystallization is also a chemical solid-liquid separation technique, in which mass transfer of a
solute from the liquid solution to a pure solid crystalline phase occurs.
Crystallization in nature
There are many examples of natural process that involve crystallization.
Usual time scale process examples include:
Process
The crystallization process consists of two major events,
nucleation and
crystal growth.
Nucleation is the step where the
solute molecules dispersed in the
solvent start to gather into clusters, on the
nanometer scale (elevating solute concentration in a small region), that becomes stable under the current operating conditions. These stable clusters constitute the nuclei. However when the clusters are not stable, they redissolve. Therefore, the clusters need to reach a critical size in order to become stable nuclei. Such critical size is dictated by the operating conditions (
temperature,
supersaturation, etc.). It is at the stage of nucleation that the atoms arrange in a defined and
periodic manner that defines the
crystal structure — note that "crystal structure" is a special term that refers to the relative arrangement of the atoms, not the macroscopic properties of the crystal (size and shape), although those are a result of the internal crystal structure.
The crystal growth is the subsequent growth of the nuclei that succeed in achieving the critical cluster size. Nucleation and growth continue to occur simultaneously while the supersaturation exists. Supersaturation is the driving force of the crystallization, hence the rate of nucleation and growth is driven by the existing supersaturation in the solution. Depending upon the conditions, either nucleation or growth may be predominant over the other, and as a result, crystals with different sizes and shapes are obtained (control of crystal size and shape constitutes one of the main challenges in industrial manufacturing, such as for pharmaceuticals). Once the supersaturation is exhausted, the solid-liquid system reaches equilibrium and the crystallization is complete, unless the operating conditions are modified from equilibrium so as to supersaturate the solution again.
Many compounds have the ability to crystallize with different
crystal structures, a phenomenon called
polymorphism. Each polymorph is in fact a different thermodynamic solid state and crystal polymorphs of the same compound exhibit different physical properties, such as dissolution rate, shape (angles between facets and facet growth rates), melting point, etc. For this reason, polymorphism is of major importance in industrial manufacture of crystalline products.
Artificial methods
For crystallization (see also
recrystallization) to occur from a solution it must be
supersaturated. This means that the solution has to contain more
solute entities (molecules or
ions) dissolved than it would contain under the equilibrium (saturated solution). This can be achieved by various methods, with 1) solution cooling, 2) addition of a second solvent to reduce the solubility of the solute (technique known as
antisolvent or drown-out), 3) chemical reaction and 4) change in pH being the most common methods used in industrial practice. Other methods, such as solvent evaporation, can also be used. The spherical crystallization has some advantages (flowability, bioavailability, ...) for the formulation of pharmaceutical drugs (see ref Nocent & al., 2
ApplicationsThere are two major groups of applications for the
artificial crystallization process:
crystal production and
purification.
Crystal production
- Macroscopic crystal production: for supply the demand of natural-like crystals with methods that "accelerate time-scale" for massive production and/or perfection.
- Tiny size crystals:
Massive production examples:
Purification
Used to improve (obtaining very pure substance) and/or verify their purity.
Crystallization separates a product from a liquid feedstream, often in extremely pure form, by cooling the feedstream or adding precipitants which lower the solubility of the desired product so that it forms crystals.
Well formed crystals are expected to be pure because each molecule or ion must fit perfectly into the lattice as it leaves the solution. Impurities would normally not fit as well in the lattice, and thus remain in solution preferentially. Hence, molecular recognition is the principle of purification in crystallization. However, there are instances when impurities incorporate into the lattice, hence, decreasing the level of purity of the final crystal product. Also, in some cases, the solvent may incorporate into the lattice forming a solvate. In addition, the solvent may be 'trapped' (in liquid state) within the crystal formed, and this phenomenon is known as inclusion.
Thermodynamic view

Low-temperature SEM magnification series for a
snow crystal. The crystals are captured, stored, and sputter coated with platinum at cryo-temperatures for imaging.
The nature of a crystallization process is governed by both thermodynamic and kinetic factors, which can make it highly variable and difficult to control. Factors such as impurity level, mixing regime, vessel design, and cooling profile can have a major impact on the size, number, and shape of crystals produced.
Now put yourself in the place of a molecule within a pure and
perfect crystal, being heated by an external source. At some sharply defined
temperature, a bell rings, you must leave your neighbours, and the complicated architecture of the crystal collapses to that of a liquid. Textbook thermodynamics says that melting occurs because the
entropy, S, gain in your system by spatial randomization of the molecules has overcome the
enthalpy, H, loss due to breaking the crystal packing forces:
Gliquid < Gsolid
This rule suffers no exceptions when the temperature is rising. By the same token, on cooling the melt, at the very same temperature the bell should ring again, and molecules should click back into the very same crystalline form. The entropy decrease due to the ordering of molecules within the system is overcompensated by the thermal randomization of the surroundings, due to the release of the heat of fusion; the entropy of the universe increases.
But liquids that behave in this way on cooling are the exception rather than the rule; in spite of the
second principle of thermodynamics, crystallization usually occurs at lower temperatures (supercooling). This can only mean that a crystal is more easily destroyed than it is formed. Similarly, it is usually much easier to dissolve a perfect crystal in a solvent than to grow again a good crystal from the resulting solution. The nucleation and growth of a crystal are under kinetic, rather than thermodynamic, control.
Equipment for crystallization
1. Tank crystallizers. Tank crystallization is an old method still used in some specialized cases. Saturated solutions, in tank crystallization, are allowed to cool in open tanks. After a period of time the mother liquid is drained and the crystals removed. Nucleation and size of crystals are difficult to control. Typically, labor costs are very high.
2.
Scraped surface crystallizers. One type of scraped surface crystallizer is the Swenson-Walker crystallizer, which consists of an open
trough 0.6 m wide with a semicircular bottom having a cooling jacket outside. A slow-speed spiral
agitator rotates and suspends the growing crystals on turning. The blades pass close to the wall and break off any deposits of crystals on the cooled wall. The product generally has a somewhat wide crystal-size distribution.
3.
Double-pipe scraped surface crystallizer. Also called a
votator, this type of crystallizer is used in crystallizing
ice cream and plasticizing
margarine. Cooling water passes in the
annular space. An internal
agitator is fitted with spring-loaded scrapers that wipe the wall and provide good heat-transfer coefficients.
4.
Circulating-liquid evaporator-crystallizer. Also called
Oslo crystallizer. Here
supersaturation is reached by evaporation. The circulating liquid is drawn by the screw pump down inside the tube side of the condensing stream heater. The heated liquid then flows into the vapor space, where flash evaporation occurs, giving some
supersaturation.The vapor leaving is condensed. The supersaturated liquid flows down the downflow tube and then up through the bed of fluidized and agitated crystals, which are growing in size. The leaving saturated liquid then goes back as a recycle stream to the
heater, where it is joined by the entering fluid. The larger crystals settle out and
slurry of crystals and mother liquid is withdrawn as a product.
5.
Circulating-magma vacuum crystallizer. The magma or suspension of crystals is circulated out of the main body through a circulating pipe by a
screw pump. The magma flows though a heater, where its temperature is raised 2-6 K. The heated
liquor then mixes with body slurry and
boiling occurs at the liquid surface. This causes
supersaturation in the swirling liquid near the surface, which deposits in the swirling suspended crystals until they leave again via the circulating pipe. The vapors leave through the top. A steam-jet
ejector provides
vacuum.
6.
Continuous oscillatory baffled crystallizer (
COBC). The COBC is a tubular baffled crystallizer that offers
plug flow under
laminar flow conditions (low flow rates) with superior heat transfer coefficient, allowing controlled cooling profiles, e.g.
linear, parabolic, discontinued, step-wise or any type, to be achieved. This gives much better control over
crystal size,
morphology and consistent crystal products. For further information see
oscillatory baffled reactor.
Filtration
Filtration is a mechanical or physical operation which is used for the separation of solids from fluids (liquids or gases) by interposing a medium through which only the fluid can pass. Oversize solids in the fluid are retained, but the separation is not complete; solids will be contaminated with some fluid and filtrate will contain fine particles (depending on the pore size and filter thickness).
Applications
* Filtration is used to separate particles and fluid in a suspension, where the fluid can be a liquid, a gas or a supercritical fluid. Depending on the application, either one or both of the components may be isolated.
* Filtration, as a physical operation is very important in chemistry for the separation of materials of different chemical composition. A solvent is chosen which dissolves one component, while not dissolving the other. By dissolving the mixture in the chosen solvent, one component will go into the solution and pass through the filter, while the other will be retained. This is one of the most important techniques used by chemists to purify compounds.
* Filtration is also important and widely used as one of the unit operations of chemical engineering. It may be simultaneously combined with other unit operations to process the feed stream, as in the biofilter, which is a combined filter and biological digestion device.
* Filtration differs from sieving, where separation occurs at a single perforated layer (a sieve). In sieving, particles that are too big to pass through the holes of the sieve are retained (see particle size distribution). In filtration, a multilayer lattice retains those particles that are unable to follow the tortuous channels of the filter. Oversize particles may form a cake layer on top of the filter and may also block the filter lattice, preventing the fluid phase from crossing the filter (blinding). Commercially, the term filter is applied to membranes where the separation lattice is so thin that the surface becomes the main zone of particle separation, even though these products might be described as sieves.
* Filtration differs from adsorption, where it is not the physical size of particles that causes separation but the effects of surface charge. Some adsorption devices containing activated charcoal and ion exchange resin are commercially called filters, although filtration is not their principal function.
* Filtration differs from removal of magnetic contaminants from fluids with magnets (typically lubrication oil, coolants and fuel oils), because there is no filter medium. Commercial devices called "magnetic filters" are sold, but the name reflects their use, not their mode of operation.
The remainder of this article focuses primarily on liquid filtration.
There are many different methods of filtration; all aim to attain the separation of substances. Separation is achieved by some form of interaction between the substance or objects to be removed and the filter. The substance that is to pass through the filter must be a fluid, i.e. a liquid or gas. Methods of filtration vary depending on the location of the targeted material, i.e. whether it is dissolved in the fluid phase or suspended as a solid.
Two main types of filter media are employed in the chemical laboratory— surface filter, a solid sieve which traps the solid particles, with or without the aid of filter paper (e.g. Büchner funnel, Belt filter, Rotary vacuum-drum filter, Crossflow filters, Screen filter), and a depth filter, a bed of granular material which retains the solid particles as it passes (e.g. sand filter). The first type allows the solid particles, i.e. the residue, to be collected intact; the second type does not permit this. However, the second type is less prone to clogging due to the greater surface area where the particles can be trapped. Also, when the solid particles are very fine, it is often cheaper and easier to discard the contaminated granules than to clean the solid sieve.
Filter media can be cleaned by rinsing with solvents or detergents. Alternatively, in engineering applications, such as swimming pool water treatment plants, they may be cleaned by backwashing. Self-cleaning screen filters utilize point-of-suction backwashing to clean the screen without interrupting system flow.
Fluids flow through a filter due to a difference in pressure - fluid flows from the high pressure side to the low pressure side of the filter, leaving some material behind. The simplest method to achieve this is by gravity and can be seen in the coffeemaker example. In the laboratory, pressure in the form of compressed air on the feed side (or vacuum on the filtrate side) may be applied to make the filtration process faster, though this may lead to clogging or the passage of fine particles. Alternatively, the liquid may flow through the filter by the force exerted by a pump, a method commonly used in industry when a reduced filtration time is important. In this case, the filter need not be mounted vertically.
Certain filter aids may be used to aid filtration. These are often incompressible diatomaceous earth or kieselguhr, which is composed primarily of silica. Also used are wood cellulose and other inert porous solids such as the cheaper and safer perlite.
These filter aids can be used in two different ways. They can be used as a precoat before the slurry is filtered. This will prevent gelatinous-type solids from plugging the filter medium and also give a clearer filtrate. They can also be added to the slurry before filtration. This increases the porosity of the cake and reduces resistance of the cake during filtration. In a rotary filter, the filter aid may be applied as a precoat; subsequently, thin slices of this layer are sliced off with the cake.
The use of filter aids is usually limited to cases where the cake is discarded or where the precipitate can be separated chemically from the filter.
Filtration is a more efficient method for the separation of mixtures than decantation, but is much more time consuming. If very small amounts of solution are involved, most of the solution may be soaked up by the filter medium.
An alternative to filtration is centrifugation — instead of filtering the mixture of solid and liquid particles, the mixture is centrifuged to force the (usually) denser solid to the bottom, where it often forms a firm cake. The liquid above can then be decanted. This method is especially useful for separating solids which do not filter well, such as gelatinous or fine particles. These solids can clog or pass through the filter, respectively.
Examples
Examples of filtration include
* The coffee filter to keep the coffee separate from the grounds
* HEPA filters in air conditioning to remove particles from air
* Belt filters to extract precious metals in mining.
* Furnaces use filtration to prevent the furnace elements from fouling with particulates.
* Pneumatic conveying systems often employ filtration to stop or slow the flow of material that is transported, through the use of a baghouse.
* In the laboratory, a Büchner funnel is often used, with a filter paper serving as the porous barrier.
An experiment to prove the existence of microscopic organisms involves the comparison of water passed through unglazed porcelain and unfiltered water. When left in sealed containers the filtered water takes longer to go foul, demonstrating that very small items (such as bacteria) can be removed from fluids by filtration.
In the kidney, renal filtration is the filtration of blood in the glomerulus, followed by selective reabsorbtion of many substances essential for the body.
References
1. Lecture notes, Postgraduate course on Filtration and Size separation at the Department of Chemical Engineering, University of Lougborough, England
2. Sterlitech
3. How does a Brita water filter work FAQ
4. Eclipse Magnetics – Magnetic filter supplier
For enquiries please send us email at
millenniumtutors@yahoo.com